4. NGS 분석 알고리즘
1) NGS 데이터 정도관리 - quality control, QC
NGS는 기술적 한계와 실험적 원인에 의한 다양한 오류(error)의 가능성이 있다. NGS 염기서열분석 결과에서는 추정 오류 확률을 수치로 나타내며 Phred 점수가 각 염기의 품질을 나타내는 지표로 활용되며 일반적으로 Q30 이상의 Phred 점수를 보이는 염기는 시퀀싱 품질이 우수하다고 판단하여 분석에 활용된다. 각 시퀀싱 리드(read)의 염기서열과 Phred 점수를 같이 표시한 것을 FASTQ 파일이라 부른다.

2) 매핑 - Mapping
FASTQ 파일의 시퀀싱 리드(read)가 어떤 염색체에 어느 위치에 있는 DNA 인지에 대한 정보를 표준 유전체(reference genome)에서 위치를 찾아주는 작업을 매핑(mapping)이라 부르며 현재 BWA 라는 프로그램이 가장 널리 이용된다. 매핑이 완료되면 각 시퀀싱 리드 별로 표준유전체에서의 염색체 번호 및 위치가 기록되는데 이것을 BAM(binary alignment map) 파일이라 부른다.

3) 변이 검출 - Variant calling
BAM 파일의 각 시퀀싱 리드를 분석하여 특정 위치에서 표준 유전체 서열과 다른 변이(variation)가 있는지 찾아내는 작업을 변이 검출(variant calling)이라 부른다. 이것은 여러 개의 시퀀싱 리드를 종합하여 확률적으로 판단하여 에러를 배제하고 진양성(true positive) 변이를 추정하는 통계적 알고리즘들이 이용되며 미국 Broad 연구소에서 개발한 GATK 프로그램이 가장 널리 사용된다. 검출된 변이는 variant call format(VCF) 형식의 파일로 저장이 되며 이 파일에는 변이의 위치와 관찰된 변이의 종류 등이 기록된다.

유전 변이는 크게 생식세포(germ-line) 및 체세포(somatic) 변이로 나뉠 수 있으며 생식세포 변이는 보통 아버지와 어머니에게서 각각 물려받은 2개의 유전자 중 하나(이형접합자, heterozygote) 혹은 2개 모두(동형접합자, homozygote)에서 변이가 관찰될 수 있기 때문에 전체 시퀀싱 리드 중 약 50% 혹은 100%로 변이가 관찰된다. 반면 체세포 변이는 후천적으로 발생하는 것으로 일부 조직 혹은 일부 세포에서만 변이가 관찰되기 때문에 변이의 비율이 1% 미만에서 99% 이상까지 다양한 비율로 관찰될 수 있다.
4) 유전자 복제수 - Copy number variation, CNV
인간의 세포는 2개의 유전자를 가지고 있으므로 대부분의 유전자 복제수(gene copy)는 2개이지만 이러한 복제 수가 늘어나거나(중복, duplication) 줄어들(결손, deletion) 수 있으며 이것을 유전자 복제수 변화(copy number variation, CNV)라 부른다. NGS 데이터에서도 복제수 또는 구조적 변화를 검출하는 데에는 여러 가지 원리의 알고리즘이 사용될 수 있다. 시퀀싱 리드의 깊이(depth of coverage)의 차이를 비교하여 중복 혹은 결손을 추정할 수 있다.

5) NGS 분석 서버 및 프로그램
NGS 원데이터(raw data)는 용량이 큰 텍스트(text) 파일의 일종으로서 분석에 많은 시간이 소요되는 만큼 이것을 쪼개서 여러 개의 CPU 코어에 할당하여 나누어 분석하도록 하는 병렬 연산(parallel computing)을 한다. 일반 분석자가 이러한 고용량 컴퓨터를 구매하고 관리하는 것이 어렵기 때문에 클라우드 컴퓨팅(cloud computing)을 이용하여 리소스를 이용하는 것도 늘고 있다. NGS 분석 프로그램 중에는 무료로 공개된 오픈소스(open-source) 프로그램과 상업적 목적으로 개발하여 유료 프로그램이 있으며, 오픈소스 프로그램은 보통 명령어 형태로 되어 있는 경우가 많아 프로그래밍에 대한 지식이 필요하다. 상용화 프로그램은 그래픽 인터페이스(graphic user interface) 기반에 자동화되어 편리한 반면 최신 알고리즘에 대한 업데이트가 느리기 때문에 사용자가 원하는 사항을 모두 만족시키기 어려운 단점이 있다.
6) 유전체 데이터베이스(database) 및 주석 (annotation)
NGS에서 변이가 검출되면 이 변이가 어느 데이터베이스에 등재되어 있는 변이인지 등을 확인해야 한다. 이 과정을 컴퓨터 알고리즘으로 자동화하는 것이 필요하며 이것을 주석(annotation)이라 부른다. 데이터베이스에는 정상인의 변이 빈도를 기록한 것이 있고(예. ExAC, gnomAD 등), 질환과 관련되어서 생식세포 돌연변이 위주로 구성된 데이터베이스(ClinVar, HGMD, LOVD 등)와 암 돌연변이 위주의 데이터베이스(COSMIC, TCGA, ICGC 등)가 있다.
7) 컴퓨터 시뮬레이션 알고리즘 - in silico
염기서열의 변화가 단백질의 구조나 기능에 어떤 영향을 줄 것인지를 컴퓨터 시뮬레이션(in silico) 알고리즘으로 예측해볼 수 있다. 그러나 이러한 알고리즘의 정확도와 특이도는 대략 60~80% 정도 되기 때문에 이 예측만을 가지고 변이의 분류에 사용하거나 임상적 결정을 내리는 것은 지양해야 한다.
8) 데이터 시각화 - visualization
NGS 데이터를 눈으로 직접 확인이 가능하게 해주는 프로그램들이 있으며, 미국 Broad 연구소에서 개발한 Integrated Genome Viewer(IGV) 프로그램이 많이 사용된다.
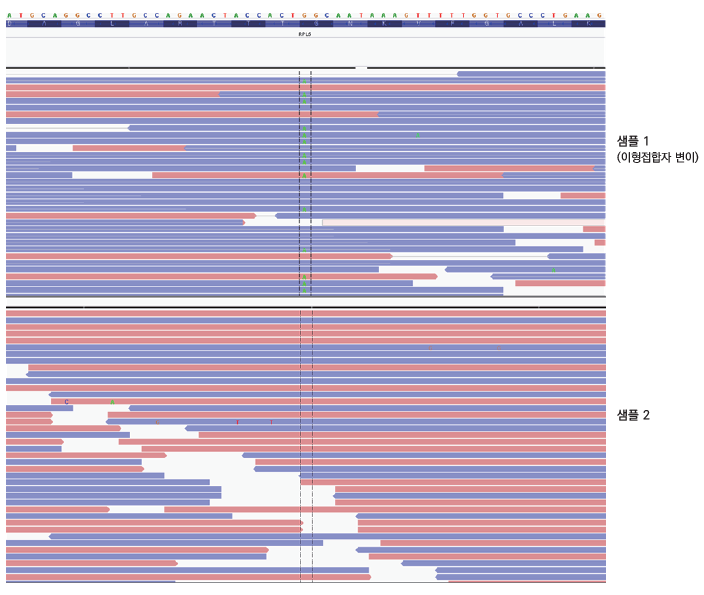
출처: 식품의약품안전처 식품의약품안전평가원 - 차세대염기서열분석 해설서
'[분자생물학] NGS' 카테고리의 다른 글
| NGS(Next Generation Sequencing) 기반 유전자 검사의 이해 - 심화용 (2) (0) | 2022.08.09 |
|---|---|
| NGS(Next Generation Sequencing) 기반 유전자 검사의 이해 - 심화용 (1) (0) | 2022.08.09 |
| NGS(Next Generation Sequencing) 기반 유전자 검사의 이해 - 입문용 (5) (0) | 2022.08.09 |
| NGS(Next Generation Sequencing) 기반 유전자 검사의 이해 - 입문용 (4) (0) | 2022.08.09 |
| NGS(Next Generation Sequencing) 기반 유전자 검사의 이해 - 입문용 (3) (0) | 2022.08.09 |



